DISCLAIMER: This blog post solely represents my own opinion based on experience, my own opinions and/or interpretations.
What’s in the name?
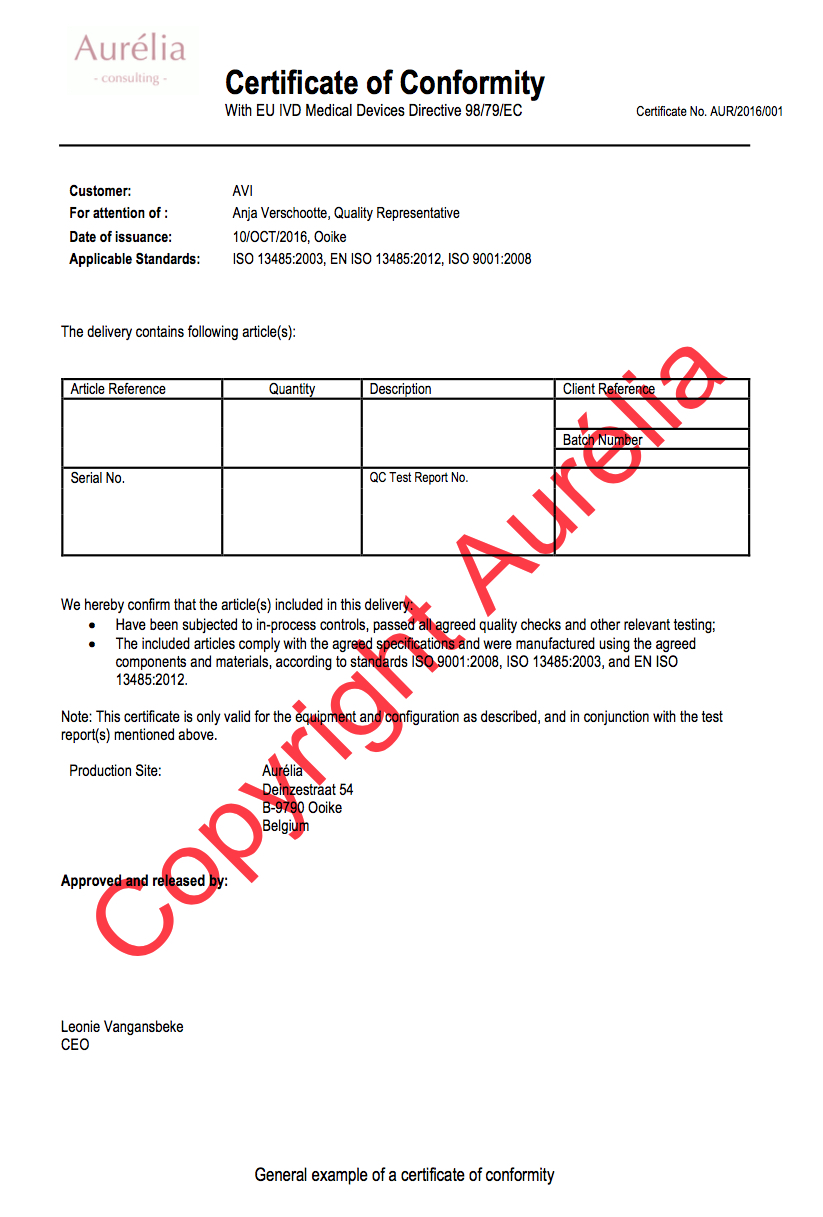
COC (Certificate of Conformity)
A certificate of Conformity or COC is a statement of a supplier which says that the product delivered meets your predefined specifications and/or the specifications of known standard(s). It’s a way of saying ‘our product was made in accordance with your required specification’
COA (Certificate of Analysis)
is a document issued by Quality Assurance that confirms that a regulated product meets its product specification. They commonly contain the actual results obtained from testing performed as part of quality control of an individual batch of a product.It’s a way of saying ‘ the material or product I’ve supplied has been analyzed to a certain standard or procedure and here are the results’.
Are they sufficient?
In my opinion, I would not solely rely on a COC as evidence that the delivered products were manufactured according to your predefined standards. An audit should be performed on the supplier or OEM to verify the quality management system and the capability to test your product throughout the manufacturing process (incoming, in-process, and finished product stages).
Search for data?
A COC provides little data for evaluation of the delivered product, it just states that the product is conforming to your specifications and/or standard’s specifications.
A COA (Certificate of Analysis) would provide more useful data. The COA includes the actual specifications agreed upon or at least the confirmation that the specifications fall within the agreed upon range. The COA also provides information about known deviations in the manufacturing process and possible contaminants.
What would the regulatory body want?
The regulatory body (FDA, FAMPH, Health Canada…) would rather see a COA than a COC. A COC is sufficient when you can provide substantive evidence that your supplier quality management process is sufficient to ensure the accuracy of the certificate.
How do you develop a COC?
Company style document
The COC should be drafted in a document that bares the company style (logo, color…).
Content
Following information should be provided but is not limited to :
- Title (e.g. Certificate of Conformity with EU IVD Medical Devices Directive 98/79/EC))
- Certificate ID;
- Customer details;
- For attention of (contact information customer, usually the QA responsible);
- Applicable standards (e.g. ISO, safety standards…);
- Article details (e.g. Article reference, quantity, description, client reference, batch ID, Serial No., Test report ID…);
- A declaration that the manufacturer complies with the predefined specifications and standards;
- Production site details;
- Approved and released by section;
- Signoff(s).
General Example COC